
Rob · @rob
791 followers · 566 posts · Server genomic.socialRT @nomad421@twitter.com
Of course, this requires mapping & quantification be performed against a single index containing both spliced & unspliced (nascent) transcripts. To this end, we suggest the piscem index (introduced at #biodata22), which can map against this enhanced transcriptome in <3GB of RAM!
Rob · @rob
791 followers · 566 posts · Server genomic.socialOf course, this requires mapping & quantification be performed against a single index containing both spliced & unspliced (nascent) transcripts. To this end, we suggest the piscem index (introduced at #biodata22), which can map against this enhanced transcriptome in <3GB of RAM!

ashish(@tp53) · @acgt01
42 followers · 595 posts · Server genomic.social
RT @ben_lengerich@twitter.com
Great presentation by @probablybots@twitter.com on contextualized networks for cancer analysis! @CSHL@twitter.com #biodata22
https://www.youtube.com/watch?v=MTcjFK-YwCw
🐦🔗: https://twitter.com/ben_lengerich/status/1602396358756925463
Rob · @rob
658 followers · 437 posts · Server genomic.socialMarkus gave an excellent talk (the last of the meeting) at #biodata22 on this work. It's very exciting, and I'm glad to see it published now. I look forward to reading the paper!

Reese Richardson · @three_body_problem
6 followers · 2 posts · Server mastodon.social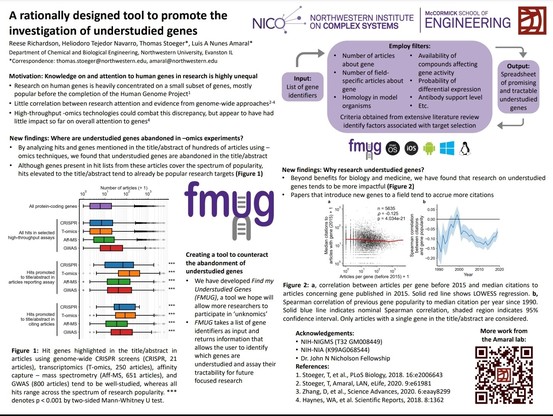
Rob · @rob
405 followers · 161 posts · Server genomic.social
RT @nomad421@twitter.com
Last Saturday at #biodata22, I talked about our recent work on spectrum preserving tilings (SPTs). The slides for that talk can be found here (https://umd.box.com/s/b0klpykjkdui5ptq34fmnz68o62bbtbn).
One key highlight is that we have made the initial releae of piscem. Why is that exciting? A short 🧵1/12

Giulio E. Pibiri :verified: · @jermp
68 followers · 28 posts · Server genomic.social
Take a moment and check Rob’s slides about SPT and the new modular indexing framework “piscem”. Still many open challenges ahead. Reach out if interested!
RT @nomad421@twitter.com
Last Saturday at #biodata22, I talked about our recent work on spectrum preserving tilings (SPTs). The slides for that talk can be found here (https://umd.box.com/s/b0klpykjkdui5ptq34fmnz68o62bbtbn).
One key highlight is that we have made the initial releae of piscem. Why is that exciting? A short 🧵1/12
Rob · @rob
401 followers · 146 posts · Server genomic.social
Last Saturday at #biodata22, I talked about our recent work on spectrum preserving tilings (SPTs). The slides for that talk can be found here (https://umd.box.com/s/b0klpykjkdui5ptq34fmnz68o62bbtbn).
One key highlight is that we have made the initial releae of piscem. Why is that exciting? A short 🧵1/12
Giulio E. Pibiri :verified: · @jermp
68 followers · 28 posts · Server genomic.social
Try the new "piscem" tool by @nomad421@twitter.com, based on the mature libraries Cuttlefish2 and SSHash!
Libraries are here:
https://github.com/COMBINE-lab/piscem-cpp
https://github.com/COMBINE-lab/cuttlefish
https://github.com/jermp/sshash
RT @holtjma@twitter.com
RT @mikelove
rob new implementation Piscem, already in use in different contexts worked on by the lab #biodata22
https://genomic.social/@mikelove/109331620896742652

Michael Love · @mikelove
661 followers · 72 posts · Server genomic.social
Short thread on my poster from #biodata22 on detecting allelic imbalance at isoform-level and in single cells, work with Rob Patro, Noor Singh, Euphy Wu et al.
PDF here: https://www.dropbox.com/s/yjr0d4mndnozwmd/DATA_22_Love.pdf?dl=0
Rob · @rob
401 followers · 146 posts · Server genomic.social
RT @mike_schatz@twitter.com
Go Katie!!!! #biodata22
🐦🔗: https://twitter.com/mike_schatz/status/1591463341234753536
Michael Love · @mikelove
661 followers · 72 posts · Server genomic.social@timtriche yeah I’ve been manually cross posting.
At #biodata22 I found Twitter was easier to use, eg I want to quickly look up handles and draft posts / threads during sessions, mostly to promote work by Phd students. Both of those are hard to do here (the latter not possible w the main app).
But for me this is week 1 of trying a new thing, entirely OSS and hosted/moderated by volunteers so I’ve got lots of patience to figure things out

Matt Holt 🦀 · @holtjma
74 followers · 80 posts · Server genomic.socialThat's a wrap on #biodata22, next one is #biodata24 on November 6-9, 2024.
Wish I could've attended in person this year, but a quick shout out to all the organizers who enabled a quick pivot to virtual! You da real MVPs!
Matt Holt 🦀 · @holtjma
74 followers · 80 posts · Server genomic.social
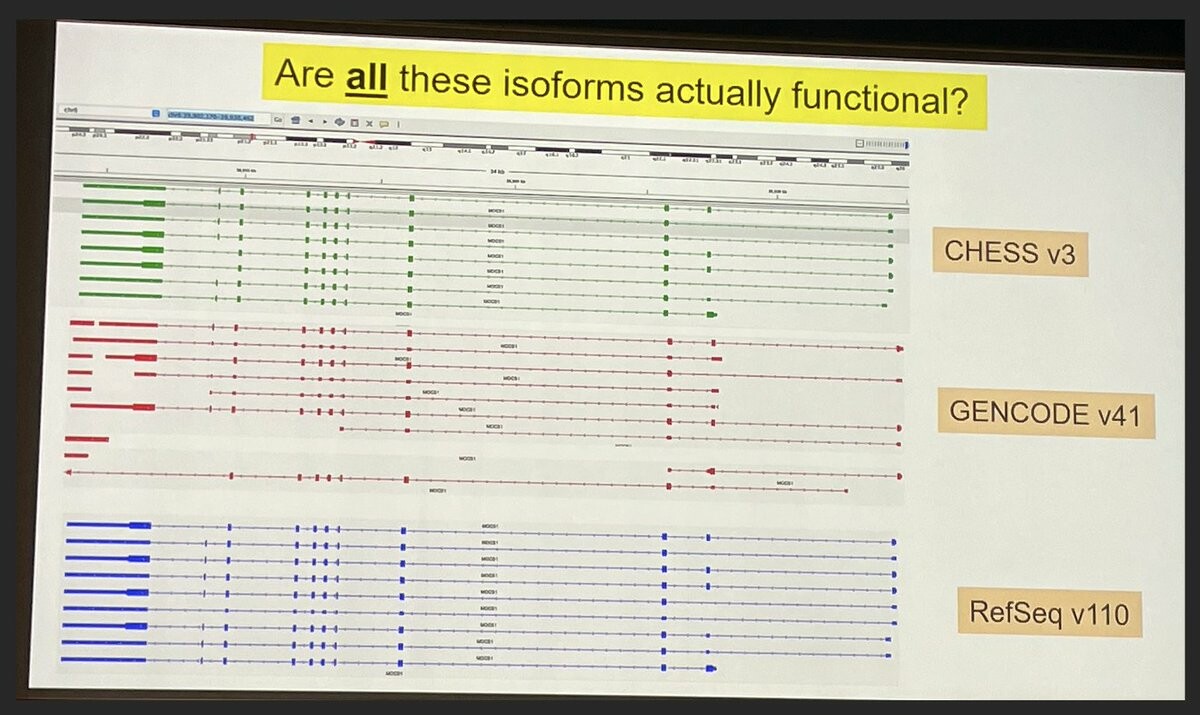
Markus Sommer #biodata22 closing us out with "Structure‐guided isoform analysis for the human transcriptome".
Problem: We have many more transcript annotations than genes. Which isoforms actually represent functional proteins?
Leveraging folding algorithms (e.g. AlphaFold2) to score each isoform, high score = more likely to be functional. Showed some examples where this scoring approach matches experimental data. Says not perfect, but helpful data point.
Website: https://www.isoform.io/
Rob · @rob
401 followers · 146 posts · Server genomic.social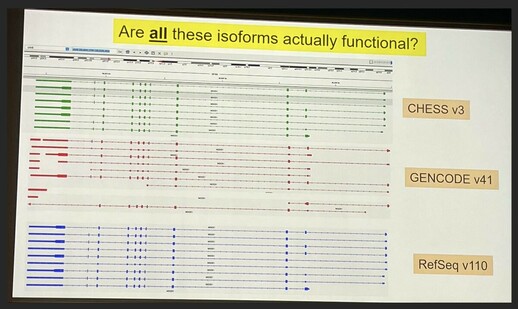
RT @mikelove@twitter.com
Last talk of #biodata22! Work by @markusjsommer@twitter.com @StevenSalzberg1@twitter.com on using protein structure information to discover transcript isoforms
Rob · @rob
401 followers · 146 posts · Server genomic.social
RT @mike_schatz@twitter.com
Closing out the conference, @markusjsommer@twitter.com will present “Structure‐guided isoform analysis for the human transcriptome” #biodata22 https://www.biorxiv.org/content/10.1101/2022.06.08.495354v1
🐦🔗: https://twitter.com/mike_schatz/status/1591472218290245633
Rob · @rob
401 followers · 146 posts · Server genomic.social
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
RT @mike_schatz@twitter.com
On now is @TBaharav@twitter.com discussing “A statistical reference‐free genomic algorithm subsumes common workflows and enables novel discovery” #biodata22 https://www.biorxiv.org/content/10.1101/2022.06.24.497555v1
🐦🔗: https://twitter.com/mike_schatz/status/1591436562491138048
Rob · @rob
401 followers · 146 posts · Server genomic.socialRT @mike_schatz@twitter.com
Up next is Jessica Bonnie describing “DandD—Utilizing “Delta delta” (Δδ) to quantify novel contributions from genomes” #biodata22 https://www.linkedin.com/in/jessicabonnie
🐦🔗: https://twitter.com/mike_schatz/status/1591442035550322690
Giulio E. Pibiri :verified: · @jermp
68 followers · 28 posts · Server genomic.socialRT @holtjma@twitter.com
Robert Patro (@rob) #biodata22 on "Keeping k‐mers in check—Building fast, small, and composable indices based on the De Bruijn graph".
Problem: Reference indexing is challenging, as we add reference (e.g. pangenome), the index grows rapidly. How do we (1/2)
Giulio E. Pibiri :verified: · @jermp
68 followers · 28 posts · Server genomic.socialRT @mike_schatz@twitter.com
Next is session co-chair @nomad421@twitter.com presenting “Keeping k‐mers in check—Building fast, small, and composable indices based on the De Bruijn graph” #biodata22 https://genomebiology.biomedcentral.com/articles/10.1186/s13059-022-02743-6
🐦🔗: https://twitter.com/mike_schatz/status/1591455428864790528